| VIBCA |
VIBrational CAlculations
|
The
core of this program was written by Patrick Fowler in the early 80's.
In 1989 it was handed to me for further development, when the range of
calculated spectroscopic observables was considerably extended.
The
program is limited to the harmonic formulation of the vibrational
problem (i.e. only the quadratic force field) and allows calculation of:
- vibrational frequencies
- eigenvectors (which can be displayed with VECTOR)
- various matrices associated with the
vibrational problem: B, G, L, and PED
(Potential Energy Distribution) matrices
- Coriolis coefficients
- quartic centrifugal distortion constants in
several reductions of the rotational Hamiltonian
- harmonic contributions to moments of inertia
- experimental values of both moments of
inertia and centrifugal distortion constants can be brought into the
calculation
VIBCA
requires input of force field in internal coordinates, but itself works
via mass-weighted Cartesian coordinate type of calculation as described
in W.D.Gwinn, J.Chem.Phys. 55, 477 (1971),
which avoids the problems inherent in the definitions of symmetry
coordinates brought in by the associated reduntant coordinates.
The
main input consists of atomic coordinates, internal coordinate
declarations and force constant values. Input deck can either be
created by hand, or most usefully, can be generated by means of program
FCONV from an appropriate ab initio calculation carried
out with the package GAMESS.
NEW: VIBCA has
been extended to deal with up to 100 atoms with associated improvements
in readability of output. The PED calculation has also been upgraded as
the previously oversimplified version was prone to some spurious
results.
|
 |
| |
| VIBCA.FOR |
The
listing - it is recommended that extension .VIB be used for the
data files |
| VIBCA.EXE |
Win32 executable (VIBCA is a straightforward console
program so that the executable can be generated with any contemporary
FORTRAN compiler) |
vibca
|
Ubuntu executable compiled with gfortran using the command:
gfortran -fno-automatic vibca.for -o vibca
|
| |
|
| MENC.VIB |
Input
file for methyl isocyanide, created from the data in J.L.Duncan et al, J.Mol.Spectrosc.
76, 55 (1979). The force field
in the paper is in symmetry coordinates, and the force constants in
internals required by VIBCA were generated with FCONV |
| MENC.RES |
Output from the above - in
comparing with the data in the paper note that you have to compare with
the (not explicitly tabulated) calculated values, such as with w-e in Table III. |
| |
|
| ANIDFT.VIB |
Data set for anisole derived with FCONV from
B3LYP/6-31G(d,p) calculation performed with PC-GAMESS |
| ANIDFT.RES |
VIBCA results file for the data set above, as reported in PCCP
7, 1708-1715 (2005); 7,
2080 (2005) |
|
|
 |
Back
to the table of programs
| FCONV |
Force Constant cONVersions
|
This
program will carry out the following conversions:
- internal to symmetry force constants
- symmetry to internal force constants
- output from a GAMESS force field run into a VIBCA
input deck
The
first two options require declaration of the U matrix and
appropriate instructions can be found at the top of the listing.
FCONV has
recently been used almost exclusively as a GAMESS to VIBCA
converter, and the H2O example below gives a complete trace
of this type of calculation: from ab inito input deck to
spectroscopic observables.
The
success of the GAMESS to VIBCA conversion depends on the declaration of internals in
GAMESS. Even though GAMESS and VIBCA recognise a common standard set of internals, such
declarations are often not straightforward, and some practice is
necessary. In general you can be certain of successful conversion only
once the calculated vibrational frequencies from GAMESS and VIBCA are
in complete agreement.
|
|
| |
| FCONV.FOR |
The
listing. Note that output will be written to two files: FC.RES and FC.VIB.
For conversion from GAMESS the
output file FC.VIB will contain a complete input deck for VIBCA,
while for symmetry<->internal force field conversions FC.VIB will contain
only a part of the necessary VIBCA deck, namely force constant values and declarations of the
potential terms
|
| FCONV.EXE |
Win32 executable (FCONV is a straightforward console
program so that the executable can be generated with any contemporary
FORTRAN compiler) |
| |
|
| |
Symmetry <-> Internal force constants |
| MENC.F |
Data for methyl
isocyanide necessary for the symmetry->internal force field conversion |
| FC1.RES |
The FC.RES file for the
above |
| |
|
| |
GAMESS to VIBCA
conversion |
| H2O.INP |
GAMESS input deck for
calculation of vibrational frequencies of water at the aug-cc-pVDZ/MP2
level.
Note that both a more accurate
vibrational calculation and its successful conversion by FCONV are
ensured by the following choice of keywords in the $FORCE group:
$FORCE NVIB=2
PURIFY=.TRUE. PRTIFC=.TRUE. VIBANL=.TRUE. DECOMP=.TRUE. $END
|
| H2O.OUT |
Abbreviated GAMESS
output for the run above |
| H2O.VIB |
VIBCA deck generated automatically by FCONV from the output
above |
| H2O.RES |
VIBCA output with H2O.VIB as input, compare frequencies with those in H2O.OUT
Try changing isotopic masses in H2O.VIB and compare
calculated quartic c.d. constants against Table 8.25 in Gordy&Cook,
while remembering that water is a particularly challenging molecule.
Note that the bottom lines predict ground state inertial defect of
0.0466, to compare with exptal value of 0.0515 uA**2.
For more rigid, heavier molecules
the experimental quartics may be expected to be reproduced/predicted to
an accuracy of about 10%. See, for example, results for pyrimidine in J.Mol.Spectrosc.
195, 332 (1999).
|
|
|
|
Back to the
table of programs
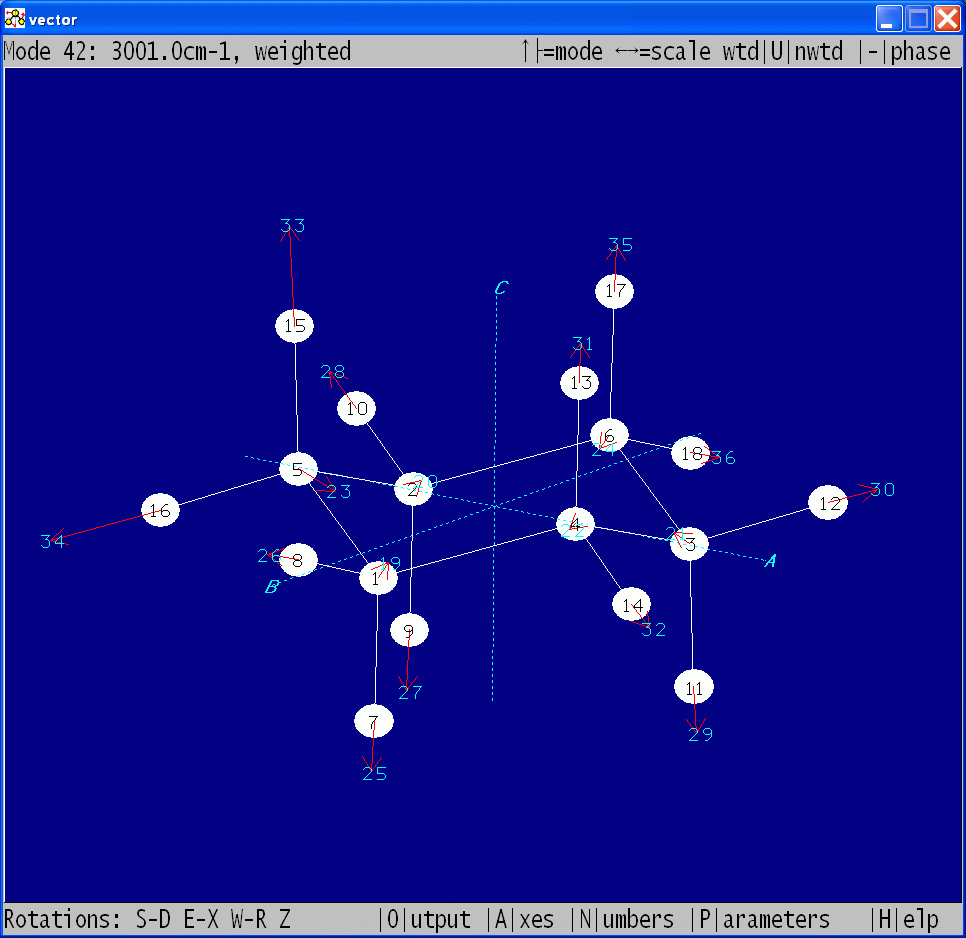
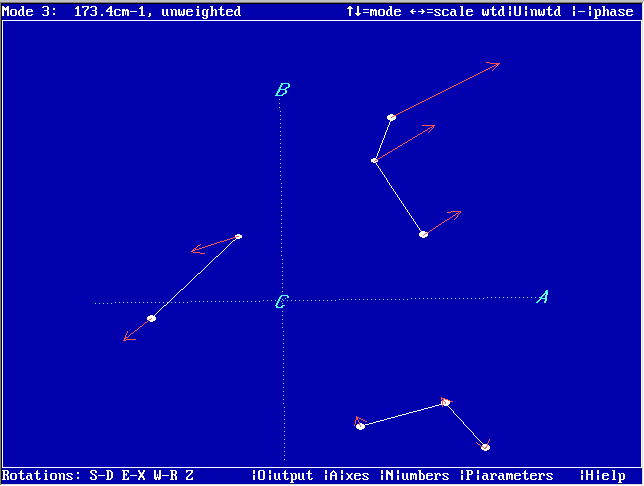
| VECTOR |
Graphical display of
normal coordinate displacement vectors calculated with VIBCA
|
This
program will graphically display eigenvectors (normal coordinate
displacement vectors) calculated with VIBCA by using the
option IFVCT=1. The program is derived from PMIFST and works similarly. The previewed eigenvectors can be:
- toggled through
- rotated
- scaled
- reversed
- plotted as mass weighted/unweighted
- output
for the gle
program, which allows generation of PostScript, PDF, JPEG etc.
publication-quality diagrams
- NEW: input also from CFOUR, GAUSSIAN, FIREFLY
The
development of VECTOR has been on standby since 2001 because there are many
excellent programs currently available for displaying normal
modes. Two examples are MOLDEN and MacMolPlt, both of
which will animate modes. On the other hand there are some
features in VECTOR that still make it useful, so that it has recently been
upgraded to be more compatible with contemporary versions of Windows
and multi-monitor desktops.
|
|
| |
| VECTOR.HDR |
Header file with basic instructions and date of the current version.
|
| VECTOR.EXE |
Executable resulting
from compilation with Intel Visual Fortran.
Key properties of the graphics, i.e.
window
size and the display font are now read from the configuration file PMIFST.CFG as used
by PMIFST.
The PMIFST.CFG file should be placed in the directory C:\ROT.
|
|
|
| VECTOR_MSF5.FOR |
Listing
of the legacy version for Microsoft Fortran 5. The issues
associated with this are discussed separately. |
| VECTOR_MSF5.EXE |
Executable from compilation with MSF5
and VGA graphics. It will run on all versions of DOS/WIN which allow
full screen MS-DOS mode. Memory requirements of this program (about 250
kB of low DOS memory) are minimal and shouldn't cause any problems. |
| |
|
| SNAP1.GIF |
Snapshot of VECTOR
screen for one of the symmetric CH stretching modes in cyclohexane (IVF
version).
|
| SNAP2.GIF |
Snapshot of VECTOR
screen for the H2O...HCl stretching mode in the (H2O)2HCl
trimer (MSF5 version). |
chex_mode1.pdf
chex_mode2.pdf
chex_mode3.pdf
|
PDF files generated from three different gle output modes available in version 2021 of VECTOR |
|
|
|
Back
to the table of programs
| ANHARM |
Energies, eigenvectors and
vibrational transitions for a reduced, one-dimensional anharmonic
potential
|
This
is a simple predictive program for the reduced anharmonic potential of
the form
V(z) = A ( c2
z2 + c3 z3 + c4
z4 + c6 z6),
which has been most often used in the reduced
quartic-quadratic form
V(z) = A ( z4
+ B z2)
for double minimum inversion potentials, in
which case B is negative. z is a dimensionless
coordinate which can be related to the molecular internal coordinate of
interest. In the quartic-quadratic form the barrier height is A B2/4,
the minima are at -B/2, and two assigned vibrational spacings
are sufficient to obtain a good idea of the potential. The matrix
elements printed by the program can also be used to set up the
dependence of various spectroscopic constants on the vibrational
quantum number. A useful description of the reduced quartic-quadratic
potential is given in J.Laane, Applied Spectroscopy 24,73-80(1970).
The
original version of this program was developed by Johan Mjöberg
and used, for example, in P.J.Mjöberg, J.Almlöf, Chem.Phys.
29,201-208(1978). In that paper the quadratic
term is positive and a cubic term is also used.
The program expects to find the data in file ANHARM.INP and
writes output to file ANHARM.OUT.
|
|
| |
| ANHARM.FOR |
The
listing |
| ANHARM.EXE |
Executable for Windows 95+ compiled with CVF6 (this is a
straightforward console application, so that it can also be compiled
with any contemporary FORTRAN compiler) |
| ANHARM2007.EXE |
Executable with corrected handling of the z6 term
|
| |
|
| ANHARM.INP |
Input file, this contains several example data
sets, the results for which can be compared against published data |
| ANHARM.OUT |
Output for thietane (trimethylene
sulfide). Check frequencies and relative intensities against Table I of
T.R.Borgers, H.L.Strauss, J.Chem.Phys. 43,947(1966). |
|
Additional information:
|
app_II.pdf
|
Tables of eigenvalues and expectation values of the
quartic-quadratic oscillator as originally produced with ANHARM (can be
compared tables in the Laane,1970 paper)
|
| chapter_2.5.pdf |
Description o the background and some molecular examples of the quartic-quadratic oscillator (see Fig.2.3)
|
|
Additional applications:
- Isotopic scaling of the reduced quartic-quadratic potential and
citations of some relevant papers: Kisiel, Krasnicki, Jabs,
Herbst, Winnewisser, Winnewisser, J.Phys.Chem A, 117, 9880-9808(2013)
- Use of ANHARM and the reduced quartic-quadratic potential for placing energy levels on an ab initio calculated potential surface: Kisiel, Pietrewicz, Fowler, Legon, Steiner, J.Phys.Chem A, 104, 6970-6978 (2000)
|
|
|
|
Back
to the table of programs
|
{kind=link}
{kind=link}